
ConfuseNN: Interpreting neural network inferences in pop gen
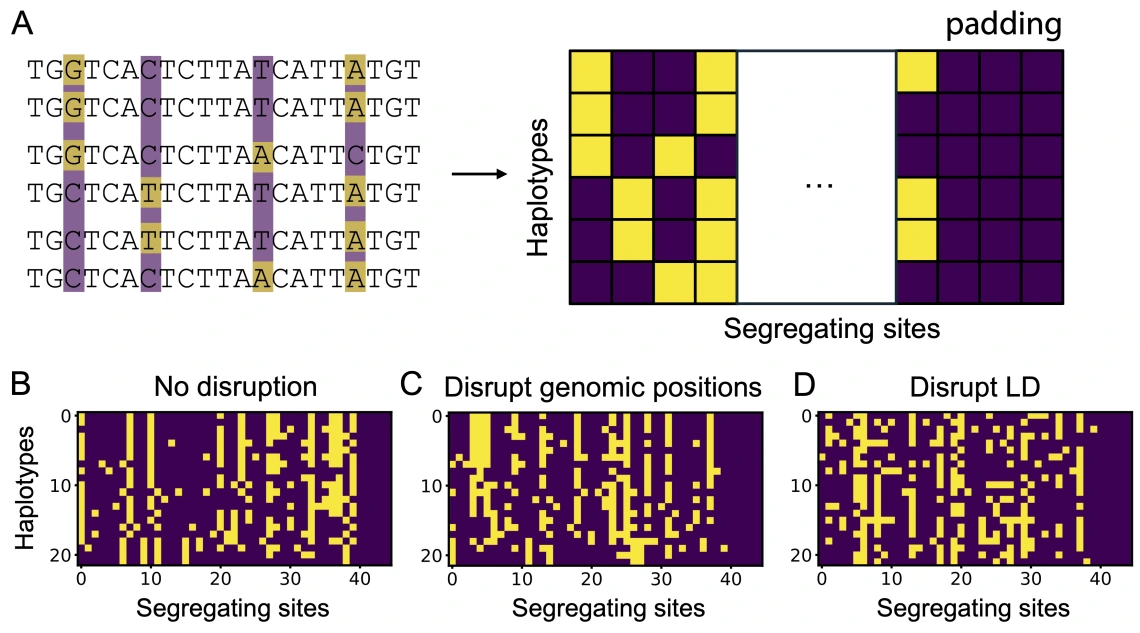
Convolutional neural networks (CNNs) have become powerful tools for population genomic inference, yet understanding which genomic features drive their performance remains challenging. Read our preprint to learn about ConfuseNN, our method for systematically shuffling input haplotype matrices to disrupt specific population genetic features and evaluate their contribution to CNN performance.
By sequentially removing signals from linkage disequilibrium, allele frequency, and other population genetic patterns in test data, we evaluate how each feature contributes to CNN performance. We applied ConfuseNN to three published CNNs for demographic history and selection inference, confirming the importance of specific data features and identifying limitations of network architecture and of simulated training and testing data design. ConfuseNN provides an accessible biologically motivated framework for interpreting CNN behavior across different tasks in population genetics, helping bridge the gap between powerful deep learning approaches and traditional population genetic theory.