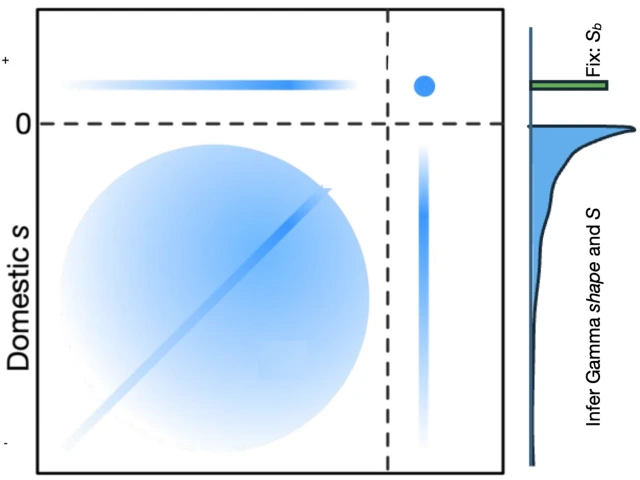
Our previous work on joint DFEs between populations only considered deleterious mutations, but adaptive mutations may be important in many cases, including domestication. In our just published collaborative paper we develop a joint DFE model that includes differential positive selection between populations and apply it to simulations designed to mimic the domestication process. The most interesting class of mutations here are those that change sign from deleterious before domestication to adaptive afterward, and we find generally good power to identify that component of the DFE if adaptation is driven by alleles of moderate effect. On the other hand, if adaptation is driven by a few alleles of strong effect, hitchhiking causes strong biases in our estimates, and those of other DFE inference tools.